
viernes, 30 de diciembre de 2016
CDHN2A (p16)
jueves, 29 de diciembre de 2016
BRC1 y BRC2

El BRCA1 y el BRCA2 son genes humanos que producen proteínas supresoras de tumores. Estas proteínas ayudan a reparar el ADN dañado y, por lo tanto, tienen el papel de asegurar la estabilidad del material genético de las células. Cuando uno de estos genes tiene una mutación, o , como cuando ya no se produce su proteína o esta no funciona correctamente, el daño al ADN no puede repararse adecuadamente. Como resultado de eso, las células tienen más probabilidad de presentar alteraciones genéticas adicionales que pueden conducir al cáncer.
Las mutaciones específicas que se heredan en el BRCA1 y en el BRCA2 aumentan el riesgo de cánceres de mama y de ovario en las mujeres y han sido asociadas con riesgos mayores de otros tipos de cáncer. Juntas, las mutaciones en el BRCA1 y en el BRCA2 representan casi de 20 a 25% de los cánceres de mama hereditarios y cerca de 5 a 10% de todos los cánceres de mama. Además, las mutaciones en el BRCA1 y en el BRCA2 representan casi 15% de los cánceres de ovarios en general. Los cánceres de mama y ovario asociados con las mutaciones en el BRCA1 y en el BRCA2 tienden a presentarse a una edad más joven que los no hereditarios.Los exámenes genéticos de BRCA1 y BRCA2 no forman parte del estudio patológico estándar. Para averiguar si tienes una anomalía genética heredada, es necesario realizar una prueba sobre una muestra de sangre.
La cartera del Servicios del SNS contempla el consejo genético en grupos de riesgo (5.3.7.1)
miércoles, 28 de diciembre de 2016
Orden de Dispensación Hospitalaria

La receta médica y las órdenes de dispensación como documentos normalizados, suponen un medio fundamental para la transmisión de información entre los profesionales sanitarios y una garantía para el paciente, que posibilita un correcto cumplimiento terapéutico y la obtención de la eficiencia máxima del tratamiento, ello sin perjuicio de su papel como soporte para la gestión y facturación de la prestación farmacéutica que reciben los usuarios del Sistema Nacional de Salud.
La orden de dispensación hospitalaria (Real Decreto 1718/2010, de 17 de diciembre, sobre receta médica y órdenes de dispensación) para pacientes no ingresados es el documento de carácter sanitario, normalizado y obligatorio para la prescripción por los médicos, odontólogos y podólogos de los servicios hospitalarios, de los medicamentos que exijan una particular vigilancia, supervisión y control, que deban ser dispensados por los servicios de farmacia hospitalaria a dichos pacientes.
Las órdenes de dispensación hospitalaria, extendidas en los hospitales públicos y privados, pueden emitirse en soporte papel, para cumplimentación manual o informatizada, y en soporte electrónico, y se editarán conforme a los criterios generales especificados en el anexo del Resal Decreto mencionado y los requisitos que las Administraciones sanitarias competentes o, en su caso, la Administración competente de las Fuerzas Armadas, introduzcan en el marco de sus competencias.
La orden de dispensación hospitalaria será dispensada por el servicio de farmacia o por el farmacéutico responsable del depósito de medicamentos del hospital en la que ha sido prescrita. En los Regímenes Especiales de las Mutualidades de Funcionarios y en el Sistema Nacional de Salud para los pacientes derivados a hospitales de referencia, podrán establecerse mecanismos que posibiliten la dispensación de la orden hospitalaria por los servicios de farmacia de los hospitales que las Administraciones competentes determinen.
La responsabilidad en la utilización de la orden de dispensación hospitalaria y la obligación de su conservación y custodia, se atendrá a lo dispuesto en el artículo 18 del real decreto para la receta médica. Una vez dispensadas, los servicios de farmacia hospitalarios conservarán las órdenes de dispensación hospitalaria, al menos durante seis meses, de acuerdo con los criterios y plazos establecidos en la Ley 41/2002, de 14 de noviembre, básica reguladora de la autonomía del paciente y de derechos y obligaciones en materia de información y documentación clínica.
El plazo de validez y los criterios de prescripción y dispensación establecidos en esta norma para la receta médica, se aplicarán a las órdenes de dispensación hospitalaria con las particularidades y características de la prestación farmacéutica en este ámbito asistencial.
En cada orden de dispensación hospitalaria se podrán prescribir uno o varios medicamentos y uno o varios envases de los mismos.
La dispensación de medicamentos se hará de acuerdo con el protocolo específico de cada tipo de tratamiento. El servicio de farmacia establecerá los mecanismos de comunicación más adecuados con los servicios médicos que permitan la atención integrada y corresponsable al paciente, comunicando al especialista prescriptor las incidencias que detecte en el seguimiento del tratamiento del paciente, con especial atención al cumplimiento y a la aparición de acontecimientos adversos. Teniendo en cuenta las especiales características de estos tratamientos, los farmacéuticos del servicio de farmacia responsables de la dispensación, podrán acceder a los datos clínicos necesarios para garantizar la efectividad y seguridad de la dispensación del medicamento.
La orden de dispensación hospitalaria electrónica se ajustará a los criterios comunes establecidos para la receta médica electrónica y, en su caso, a los específicos del Sistema Nacional de Salud, con la adaptación correspondiente al ámbito hospitalario. Para posibilitar la dispensación de los tratamientos en el supuesto contemplado en el artículo 17.2 de este real decreto, se implantará la interoperabilidad del sistema de orden de dispensación hospitalaria electrónica, según lo establecido en el artículo 7.2.
Las órdenes de dispensación hospitalaria utilizadas en el ámbito hospitalario para pacientes no ingresados, se adaptarán a lo dispuesto para la receta médica con las salvedades correspondientes, como son los espacios destinados a: Régimen de uso, y cupones precinto o asimilados, que no serán incluidos en el documento, así como para posibilitar la prescripción de varios medicamentos.
En el espacio «Contingencia», se hará constar Orden de dispensación hospitalaria.
Además, en la Orden de dispensación hospitalaria, se especificará el servicio de farmacia en lugar de la oficina de farmacia, incluyendo asimismo, el número de Historia Clínica en los datos del paciente y el servicio médico o unidad clínica, además del prescriptor.
Como comprobante de la dispensación, constará la fecha del recibí del paciente y su firma.
Por los hospitales, en la asistencia sanitaria privada, así como por las Administraciones y Organismos competentes, en caso de estimarlo oportuno, podrán incluirse en las órdenes de dispensación hospitalaria las referencias o datos y las copias necesarias para su correcta utilización y control.
La orden de dispensación, con carácter general, se atendrá a los criterios básicos establecidos en este anexo para la receta médica y se adecuará a las características que se describen en el correspondiente modelo.
En particular, la orden de dispensación oficial del Sistema Nacional de Salud incluida la de las Mutualidades de Funcionarios se atendrá a los criterios básicos establecidos en el punto primero, mientras que la orden de dispensación para la asistencia sanitaria privada deberá atenerse a los criterios básicos establecidos en el punto segundo.
jueves, 22 de diciembre de 2016
Celiaquia. Clasificación de Marsh

Para llegar a un diagnóstico es necesario conocer “la regla de los cinco dedos”, con tener cuatro, podemos diagnosticar. Es decir, hay que estudiar cinco factores compatibles, a saber: Clínica sugestiva de celiaquía, con o sin otras manifestaciones extra-digestivas; Serología, que puede ser positiva o negativa; Genética, que puede ser completa (homozigota) o incompleta (heterozigota); Resultado de las biopsias duodenales, que suelen mostrar un estadio de Marsh 1, como lesión más habitual; Respuesta a la dieta sin gluten, seguida estrictamente durante un tiempo mínimo de 6 meses. Durante y a la finalización de los seis meses de dieta sin gluten estricta suele producirse una clara mejoría clínica, aunque la inflamación intestinal es más lenta y tarda más en desaparecer por completo, pudiendo pasar hasta dos años en recuperarse de las lesiones.
En la celiaquía no hay grados. Una persona es celiaca o no es celiaca, pero nadie es más celiaco que el de al lado.
Cuando se habla de grados, se habla de grados de lesión del intestino. El daño que nuestro sistema inmunológico provoca a los celiacos es la atrofia de las vellosidades intestinales. Y en función de ese daño hablamos de grados de lesión del intestino.
Clasificación de Marsh

lunes, 19 de diciembre de 2016
Signo de Crowe

Este signo médico fue nombrado en honor del Dr. Frank W. Crowe, un médico estadounidense, quien notó que las pecas axilares están presentes en aproximadamente el 20-30% de los pacientes con neurofibromatosis, pero no veía ninguna en pacientes que no tenían neurofibromatosis.
El Instituto Nacional de la Salud de EE.UU (NIH, definió en 1987, los criterios diagnósticos de la neurofibromatosis tipo I. El signo de Crowe forma parte de estos criterios.
domingo, 18 de diciembre de 2016
Queratosis seborreica

Para su diagnóstico suele bastar con el diagnóstico clínico (lesiones cerosas, de color amarillo o marrón de forma redondeada u ovalada, con apariencia de estar adheridas, únicas aunque con tendencia a ser múltiples). En ocasiones también se puede recurrir a una biopsia de lesión de piel para confirmar el diagnóstico y descartar la presencia de un melanoma o de un epitelioma basocelular.
Este tipo de lesiones se pueden eliminar mediante cirugía, láser o crioterapia. La queratosis seborreica es benigna y por lo general indolora. Eliminar la lesión conlleva un procedimiento muy simple que no deja cicatrices. Por lo general, no vuelven a aparecer, pero las personas propensas pueden desarrollar nuevos crecimientos de este tipo con posterioridad.
viernes, 9 de diciembre de 2016
Alergia acuagénica

Se cree que es debida a la presencia en la piel de un antígeno -sustancia que activa el sistema inmune- hidrosoluble. En contacto con el agua, el antígeno se disuelve, atraviesa la piel y hace que las células de defensa liberen histamina. Esta provoca la aparición de ronchas, picor y otros síntomas alérgicos.
No hay ningún tratamiento para este mal, pero los dermatólogos recomiendan mantenerse tan lejos del agua como le sea posible, darse duchas cortas y con agua fría, evitar la lluvia y estar siempre en lugares refrigerados, porque hasta el sudor es peligroso.
Se recomienda:
- Evitar al máximo el contacto con el agua.
- Bañarse con alguna prenda de algodón ligera y reducir al máximo el tiempo de exposición.
- El tratamiento de la urticaria acuagénica incluye aplicar alguna crema para calmar la piel irritada, por lo que el paciente debe procurar tener siempre a la mano cremas o pomadas humectantes.
- En algunos casos se ha comprobado que la aplicación previa de aceite, gel o lanolina sobre la piel permite al individuo realizar deportes acuáticos, ya que reduce la posibilidad de sufrir síntomas de urticaria acuagénica.
- Es posible que este tipo de alergias raras pueda originarse por los minerales añadidos al agua, por ello, tener filtros purificadores en casa puede ser una solución.
- Consumir otros líquidos para hidratarse, por ejemplo, jugos de frutas y/o verduras e, incluso, refrescos.
- Utilizar medicamentos antihistamínicos para controlar los síntomas de alergia al agua, aunque debe recordarse que solamente se obtiene alivio temporal, pues la urticaria acuagénica no se cura.
- Es importante señalar que las molestias suelen ser temporales, es decir, la irritación al contacto con el agua desaparece por sí sola, permitiendo al paciente retomar sus actividades sin mayor problema.
miércoles, 7 de diciembre de 2016
Cirugía robótica Da Vinci

La historia
del desarrollo del Da Vinci —el robot más extendido en los hospitales
de todo el mundo— es curiosa. Comienza cuando, a finales de los años 80,
ingenieros de la NASA que trabajaban en el campo de la realidad virtual
se interesan en usar sus conocimientos para poder hacer cirugías a
distancia. Cuando, a mediados de los 90, tienen los primeros prototipos,
varios cirujanos y endoscopistas se unen al equipo, conscientes de que
podrían mejorar las por entonces menos desarrolladas técnicas de
laparoscopia.
Poco después, consciente de esa unión, la Armada de los Estados Unidos empieza a colaborar en la financiación del proyecto. “Su idea era poder operar a los heridos a distancia, por ejemplo con el paciente en un portaaviones y el cirujano en el hospital”, comenta Alcaraz. Varios de los cirujanos e ingenieros implicados en el proceso formaron diversas empresas que terminaron dando lugar al desarrollo del Da Vinci, cuyo uso fue aprobado por las autoridades sanitarias de Estados Unidos en el año 2000. En el 2001, cirujanos en Nueva York extirparon con él la vesícula de una paciente en Estrasburgo, a 6.000 kilómetros de distancia.
La Fundación Puigvert fue el primer hospital de España en contar con el robot da Vinci en su centro.
Este tipo de cirugía se lleva a cabo con instrumentos especialmente diseñados, de reducido tamaño y una gran precisión en su manejo. Este avanzado equipamiento permite trabajar con una perfecta visualización de la zona y confiere una mayor precisión al cirujano.Poco después, consciente de esa unión, la Armada de los Estados Unidos empieza a colaborar en la financiación del proyecto. “Su idea era poder operar a los heridos a distancia, por ejemplo con el paciente en un portaaviones y el cirujano en el hospital”, comenta Alcaraz. Varios de los cirujanos e ingenieros implicados en el proceso formaron diversas empresas que terminaron dando lugar al desarrollo del Da Vinci, cuyo uso fue aprobado por las autoridades sanitarias de Estados Unidos en el año 2000. En el 2001, cirujanos en Nueva York extirparon con él la vesícula de una paciente en Estrasburgo, a 6.000 kilómetros de distancia.
La Fundación Puigvert fue el primer hospital de España en contar con el robot da Vinci en su centro.
En España ya son alrededor de 25 los hospitales que han incorporado esta tecnología. Sirva como ejemplo el País Vasco que cuenta con cuatro robots en la Sanidad Pública (Osakidetza): Hospital de Donostia, Basurto, Cruces y el Hospital de Araba. En Cataluña, el Hospital Vall d´Hebron con dos quirófanos dotados de Da Vinci.
El Hospital San Carlos fue el primer hospital del Sistema Nacional de Salud en contar con el robot Da Vinci, gracias a la Fundación Esther Koplowitz (ver video).
domingo, 27 de noviembre de 2016
Reumatismo palindrómico

Es una enfermedad articular inflamatoria caracterizada por episodios breves y recurrentes de artritis, autolimitado y que no deja secuelas.
La mitad de los individuos con reumatismo palindrómico eventualmente desarrollan artritis reumatoide (AR), la cual sí genera daño perdurable a las articulaciones. Se desconocen sus causas.
El cuadro clásico de este síndrome consiste en la aparición súbita de un cuadro de artritis o periartritis afebril, que puede durar horas o días y desaparece de la misma manera en que apreció, súbitamente. Hay durante el episodio dolor de moderada a gran intensidad, rubicundez, tumefacción y calor en la o las articulaciones comprometidas. El intervalo entre los ataques es extremadamente variable, de días a meses o años. Como dijimos no deja secuelas clínicas ni radiológicas.
Las articulaciones más frecuentemente afectadas suelen ser las metacarpofalángicas, interfalángicas proximales, muñecas, rodillas y hombros, por este orden, aunque puede afectarse cualquiera.
El factor reumatoide suele ser negativo, así como los ANA (anticuerpos antinuecleares), y no se demuestra relación con antígenos HLA (antigenos leucocitarios humanos, por sus siglas en inglés), por lo que el diagnóstico es eminentemente clínico.
Afecta a hombres y a mujeres entre los 20 y los 50 años.
El uso de los antiinflamatorios no esteroideos tiene indicación sintomática. En la profilaxis de los episodios se han empleado diferentes medicamentos como los corticosteroides, la colchicina, las sales de oro o algunos antipalúdicos.
viernes, 25 de noviembre de 2016
Mutación del factor V de Leiden y mutación en el gen de la protrombina

Entre las trombofilias heredadas más importantes por su frecuencia en la población mundial se encuentran la generada por la mutación del factor V de la coagulación (denominado factor V de Leiden), y la mutación en el gen de la protrombina (factor II de la coagulación) en posición G20210A, descubierto en 1996.
Ambas mutaciones están asociadas a un mayor riesgo de trombosis.
El conocimiento de estas mutaciones han permitido elaborar una terapia definida y evitar los riesgos conocidos de trombosis para prevenir el evento trombótico
martes, 22 de noviembre de 2016
Urticaria cronica espontánea

En 1965 el Dr. SR Halpern, pediatra americano, escribió: «La urticaria crónica resulta una situación muy molesta para el paciente y para el médico. En el primero causa incomodidad, pica y es estéticamente desagradable. Mientras que para el médico es desconcertante, y consecuentemente incomoda su estabilidad intelectual porque este se ve incapaz de manejar el problema [...]».
La urticaria crónica espontánea, llamada también UCE, es una patología crónica caracterizada por la aparición espontánea de habones con o sin angioedema, que persisten al menos durante seis semanas bien diariamente o bien de forma recurrente varias veces por semana.
Del 0,5 al 1 % de los europeos están afectados por este tipo de urticaria, siendo más frecuentes entre las mujeres (relación 2:1).
Aunque puede presentarse a cualquier edad las personas de entre 20 a 40 años son las más afectadas.
Del 0,5 al 1 % de los europeos están afectados por este tipo de urticaria, siendo más frecuentes entre las mujeres (relación 2:1).
Aunque puede presentarse a cualquier edad las personas de entre 20 a 40 años son las más afectadas.
En la etiopatogenia de la urticaria crónica se han implicado infecciones, mecanismos autorreactivos/autoinmunitarios, así como reacciones "seudoalérgicas" tanto con alimentos como con fármacos. No obstante, en un grupo importante de pacientes la etiología es desconocida y es la que denominamos urticaria crónica idiopática.
La mayoría de pruebas de cribado que se realizan en los pacientes con esta afección no deben recomendarse de forma generalizada por su baja relación respecto a coste-efectividad.
Para el diagnóstico de esta urticaria resultará fundamental una historia clínica detallada , un análisis de sangre completo y en aquellos casos que se desee decartar la forma autorrecativa/autoinmunitaria se realizará el ASST (test con suero autólogo o autotest). Si da positivo habría que realizar pruebas in vitro de liberación de histamina o bien la detección de marcadores de activación de basófilos (CD63 y/o CD203c) así como demostrar la presencia de autoanticuerpos anti-IgE para el diagnóstico definitivo de urticaria crónica autoinmunitaria.
El tratamiento consiste, en primer lugar, en la identificación y eliminación de los posibles factores desencadenantes o, al menos, en su reducción significativa. En casos de urticaria activa se deben evitar el estrés, el alcohol y el calor, o el ejercicio intenso, que pueden dar lugar a exacerbaciones. Tambien los medicamentos que empeoren la situación como los AINE y los mórficos. La información al paciente en todo momento resulta fundamental.
Se utilizan como primer escalón los antihistaminicos H1 (anti-H1) no sedantes de segunda generación a dosis stándard. En el caso de que los síntomas no se controlen con los medicamentos anteriores tras dos semanas de tratamiento se recomienda aumentar las dosis de los H1 y si esto tampoco produce un efecto beneficios tras cuatro semanas de tratamiento se puede añadir un inhibidor de receptor de los leucotrienos. En cualquier momento podemos utilizar ciclos cortos de glucocorticoides si se produce exacerbación de los síntomas. En el caso de no obtener mejoría, y como tercer escalón, se pueden utilizar tratamientos con una indicación que no está en la ficha técnica (tratamientos off label) que basicamente son tres: antihistaminicos H2, inhibidores de la calcineurina (ciclosporina, tacrolimus) y omalizumab. Generalmente los subsidiarios de este tercer escalón son los pacientes diagnosticados de urticaria crónica autoinmunitaria.
El tratamiento consiste, en primer lugar, en la identificación y eliminación de los posibles factores desencadenantes o, al menos, en su reducción significativa. En casos de urticaria activa se deben evitar el estrés, el alcohol y el calor, o el ejercicio intenso, que pueden dar lugar a exacerbaciones. Tambien los medicamentos que empeoren la situación como los AINE y los mórficos. La información al paciente en todo momento resulta fundamental.
Se utilizan como primer escalón los antihistaminicos H1 (anti-H1) no sedantes de segunda generación a dosis stándard. En el caso de que los síntomas no se controlen con los medicamentos anteriores tras dos semanas de tratamiento se recomienda aumentar las dosis de los H1 y si esto tampoco produce un efecto beneficios tras cuatro semanas de tratamiento se puede añadir un inhibidor de receptor de los leucotrienos. En cualquier momento podemos utilizar ciclos cortos de glucocorticoides si se produce exacerbación de los síntomas. En el caso de no obtener mejoría, y como tercer escalón, se pueden utilizar tratamientos con una indicación que no está en la ficha técnica (tratamientos off label) que basicamente son tres: antihistaminicos H2, inhibidores de la calcineurina (ciclosporina, tacrolimus) y omalizumab. Generalmente los subsidiarios de este tercer escalón son los pacientes diagnosticados de urticaria crónica autoinmunitaria.
lunes, 21 de noviembre de 2016
Síndrome de Rett

La causa del síndrome de Rett es una alteración (mutación) en el gen MECP2 (methyl-CpG-binding protein 2), localizado en el locus (posición de un gen dentro de un cromosoma) q28 del Cromosoma X. A través de este gen se produce una proteína (llamada MeCP2) que está ampliamente distribuida a nivel del núcleo de las células y que es especialmente abundante en las neuronas maduras del sistema nervioso central. Esta proteína juega un papel importante en la regulación de la sinapsis (comunicación entre las neuronas) y es fundamental en el desarrollo del sistema nervioso tras los primeros meses de vida. Existen más de doscientas mutaciones descritas en este gen, sin embargo no se sabe con exactitud cómo actúa la proteína anormal que codifica el gen mutado para producir la enfermedad.
Aunque el síndrome de Rett (ver vídeo) es de causa genética, en cerca de un noventa y nueve por ciento de los casos la mutación aparece en el paciente espontáneamente durante su desarrollo embrionario en el vientre materno, por lo tanto no lo ha heredado de sus padres. En el uno por ciento de las familias restantes con Rett el gen defectuoso se ha transmitido desde las mujeres portadoras de la mutación hacia sus hijos. Los científicos aún están tratando de comprender el proceso de la herencia en esta complicada enfermedad.En relación a su prevalencia, esta enfermedad se clasifica en el grupo de enfermedades raras y se presenta en uno de cada diez mil recién nacidos vivos del sexo femenino, siendo la segunda causa más frecuente de retraso mental en este sexo.
Usualmente se diagnostica durante los dos primeros años de vida, lo que es fundamental para indicar tratamientos dirigidos a mejorar en retraso psicomotor que presentan los pacientes, ya que los cambios en los patrones normales de desarrollo mental y social comienzan entre los seis y los 18 meses.
Cuando la mutación aparece en recién nacidos del sexo masculino la enfermedad suele ser muy agresiva, porque los niños poseen un solo Cromosoma X y éstos mueren durante los primeros días de vida.
Se recomienda consultar con un neurólogo pediátrico o a un pediatra especializado en el desarrollo para confirmar el diagnóstico clínico del síndrome de Rett. El médico utiliza unas pautas sumamente específicas, las cuales se dividen en tres tipos de criterios clínicos diversos: esencial, de apoyo y de exclusión. La presencia de cualquiera de los criterios de exclusión niega un diagnóstico "clásico" o "típico" del síndrome de Rett.
Ejemplos de criterios o síntomas de diagnóstico esenciales incluyen un desarrollo aparentemente normal hasta los 6 y 18 meses de edad y poseer una circunferencia de la cabeza normal al nacer seguida por retrasos en el índice del crecimiento de la cabeza con el pasar del tiempo (entre los 3 meses y los 4 años de edad). Otros criterios de diagnóstico esenciales incluyen un deterioro significativo del habla, movimientos repetitivos de la mano, sacudidas del torso, caminar sobre las puntas del pie o un paso inestable, rígido y con aumento en la base de sustentación (piernas separadas).
Los criterios de apoyo no se requieren para un diagnóstico del síndrome de Rett pero pueden ocurrir en algunos pacientes. Además, estos síntomas - cuya gravedad varía de niño a niño - pueden que no estén presentes en niñas muy pequeñas, pero podrían desarrollarse con la edad. Un niño que cumpla con los criterios de apoyo pero que no cumpla ninguno de los criterios esenciales no padece del síndrome de Rett. Los criterios de apoyo incluyen dificultades en la respiración; anormalidades en el electroencefalograma (EEG); convulsiones; rigidez muscular, espasticidad y/o contracturas de las coyunturas que empeoran con la edad; escoliosis; rechinar o crujir los dientes; pies pequeños en relación a la estatura; retrasos en el crecimiento; disminución del tejido graso del cuerpo y la masa muscular (aunque puede haber una tendencia hacia la obesidad en algunos adultos afectados); patrones anormales del sueño, irritabilidad o agitación; dificultades para masticar o deglutir (tragar); mala circulación en las extremidades inferiores, con pies y piernas fríos y amoratados; movilidad disminuida con la edad; y estreñimiento.
Además de los criterios de diagnóstico esenciales, un número de condiciones específicas permite a los médicos eliminar un diagnóstico del síndrome de Rett. A estos se les conoce como criterios de exclusión. Los niños que cumplen con alguno de los criterios siguientes no padecen del síndrome de Rett: recrecimiento de los órganos del cuerpo u otras muestras de la enfermedad de almacenamiento, pérdida de la visión debido a un trastorno de la retina o atrofias ópticas, microcefalia congénita, trastornos metabólicos identificables u otros trastornos degenerativos congénitos, trastornos neurológicos adquiridos por una infección o trauma severos de la cabeza, evidencia de un retraso en el crecimiento en el útero, o evidencia de daños cerebrales adquiridos después del nacimiento.
No existe cura para este síndrome. El tramiento es sintomático y de apoyo.
Pueden requerirse medicamentos para controlar las irregularidades respiratorias y las dificultades motoras y se pueden utilizar drogas antiepilépticas para controlar las convulsiones. Debe haber supervisión regular para la escoliosis y las posibles anormalidades del corazón. La terapia ocupacional (en la cual los terapistas ayudan a los niños a desarrollar las capacidades necesarias para realizar actividades autónomas- ocupaciones -por ejemplo, vestirse, alimentarse y realizar labores artísticas o artesanales), la fisioterapia y la hidroterapia pueden prolongar la movilidad. Algunos niños pueden requerir equipo y ayuda especiales, tales como soportes para detener la escoliosis, férulas o tablillas para modificar los movimientos de la mano y programas alimenticios para ayudarles a mantener el peso adecuado. También se pueden requerir en algunos casos servicios especiales académicos, sociales, vocacionales y de ayuda.
Leer más sobre actualización clínica, diagnóstica y moleculas.
viernes, 18 de noviembre de 2016
DIARREA DEL VIAJERO (TRAVELERS’ DIARRHEA)

La costa de Marsella también goza de su serie de extraordinarias esculturas: son "Les Voyageurs", una creación en bronce del artista galo Bruno Catalano. Las obras representan un grupo de trabajadores con gran parte de sus cuerpos desaparecidos, y fueron puestas en exhibición en 2013 con motivo de que la ciudad francesa fue declarada la Capital Europea de la Cultura.
La diarrea de los viajeros (TD) es la enfermedad más frecuente relacionada con los viajes. Se calcula que entre el 30% y el 70% de los viajeros, dependiendo del destino y la temporada del viaje sufre esta afección. Tradicionalmente, se pensaba que la TD podría ser prevenida siguiendo simples recomendaciones como "hervir el agua, cocinar adecuadamente la comida, pelar las frutas ...", pero los estudios realizados con esta materia han descubierto que las personas que siguen estas reglas pueden seguir enfermándose. La escasa higiene en los restaurantes locales es probablemente el mayor riesgo de TD.
La TD es un síndrome clínico que puede surgir como consecuencia la acción de patógenos intestinales. Los patógenos bacterianos son el riesgo predominante, representando un 80% -90% de TD. Los virus intestinales suelen representar entre el 5% y el 8% de las enfermedades, aunque al mejorar el diagnóstico puede aumentar en el futuro el porcentaje de las infecciones por norovirus. Las infecciones con patógenos protozoarios son más tardías en la presentación sintomática representando aproximadamente el 10% de los diagnósticos en personas con viajes más prolongados.
Lo que comúnmente se conoce como "intoxicación alimentaria" implica la ingestión de toxinas preformadas en los alimentos. En este síndrome, los vómitos y la diarrea pueden estar presentes, pero los síntomas suelen desaparecer espontáneamente dentro de las 12 horas.
La TD es un síndrome clínico que puede surgir como consecuencia la acción de patógenos intestinales. Los patógenos bacterianos son el riesgo predominante, representando un 80% -90% de TD. Los virus intestinales suelen representar entre el 5% y el 8% de las enfermedades, aunque al mejorar el diagnóstico puede aumentar en el futuro el porcentaje de las infecciones por norovirus. Las infecciones con patógenos protozoarios son más tardías en la presentación sintomática representando aproximadamente el 10% de los diagnósticos en personas con viajes más prolongados.
Lo que comúnmente se conoce como "intoxicación alimentaria" implica la ingestión de toxinas preformadas en los alimentos. En este síndrome, los vómitos y la diarrea pueden estar presentes, pero los síntomas suelen desaparecer espontáneamente dentro de las 12 horas.
jueves, 17 de noviembre de 2016
Phadiatop y Phadiatop infant

Son 2 test cualitativos utilizados a modo de screening para detectar la presencia o la ausencia de IgE específica sérica a una variedad de alergenos inhalantes (Phadiatop®) e inhalantes + alimentarios (Phadiatop infant®) en pacientes con sospecha de enfermedades alérgicas.
El Phadiatop está indicado en caso de no disponer o estar contraindicada la realización de un prick test, siendo su principal utilidad en el ámbito ambulatorio de la medicina primaria.
El primero de estos test se compone de una mezcla de neumoalergenos (ácaros, gato, perro, gramíneas, árboles, hongos) y es útil en niños con dermatitis atópica, sibilancias recurrentes o asma.
El segundo contiene una mezcla de neumoalergenos y alérgenos alimentarios (leche, huevo, cacahuete, soja, gamba) y es útil en el estudio de menores de 4 años.
El Phadiatop está indicado en caso de no disponer o estar contraindicada la realización de un prick test, siendo su principal utilidad en el ámbito ambulatorio de la medicina primaria.
El primero de estos test se compone de una mezcla de neumoalergenos (ácaros, gato, perro, gramíneas, árboles, hongos) y es útil en niños con dermatitis atópica, sibilancias recurrentes o asma.
El segundo contiene una mezcla de neumoalergenos y alérgenos alimentarios (leche, huevo, cacahuete, soja, gamba) y es útil en el estudio de menores de 4 años.
miércoles, 16 de noviembre de 2016
Síndrome de Ulises

El síndrome de Ulises se caracteriza, por un lado, porque la persona padece unos determinados factores estresantes o duelos y, por otro, porque aparecen un amplio conjunto de síntomas psíquicos y somáticos (depresión, ansiedad, somatizaciones, etc.). En la actualidad constituye un problema de salud mental emergente en los países de acogida de inmigrantes, para el que los médicos de familia españoles debemos estar preparados como consecuencia de la última oleada de inmigración a Europa.
Entre el 15% y el 2% de los inmigrantes que visitan los servicios de salud mental en España no padecen ninguna enfermedad sino lo que padecen es el denominado "Síndrome de Ulises", que no debe ser confundido con una depresión ni con ningún tipo de enfermedad mental, sino como una manifestación específica de estrés crónico múltiple.
Los síntomas que caracterizan este síndrome son depresivos (tristeza y llanto), pensamientos de muerte y obsesivos por los "gravísimos problemas que tiene" ansiedad, irritabilidad con menores, problemas somáticos (cefaleas y fatigas por su dolor psíquico) o síntomas confusionales (pérdida de memoria).
La mayor parte de estos pacientes mejora si se les proporciona el apoyo sanitario y social necesario. La intervención incluye técnicas psicodinámicas y cognitivas, ya que el estrés hace que se produzcan ciertos errores en el procesamiento de la información o generalizaciones excesivas. También es frecuente el tratamiento farmacológico, bien con ansiolíticos o antidepresivos
Esta intervención debe completarse con un sólido apoyo psicosocial, poniendo en contacto a estos inmigrantes con asesores legales, asociaciones de inmigrantes, redes sociales que faciliten el aprendizaje del idioma y les eduquen sobre buenos hábitos de salud para mejorar su calidad de vida y contener el estrés, como son la higiene del sueño o la práctica deportiva.
lunes, 14 de noviembre de 2016
Visados de recetas médicas

Los visados de recetas de medicamentos -o, técnicamente denominados visados previos de inspección- son un mecanismo de control a través del cual la Administración Pública sanitaria lleva a cabo un control individualizado sobre la prescripción médica de unas determinadas especialidades farmacéuticas y que se impone como requisito necesario para la dispensación farmacéutica de las mismas.
La obtención de medicamentos con cargo a las cuentas públicas se encuentra fuertemente intervenida por razones sanitarias y por razones socio-económicas.
La presencia del derecho a la protección de la salud (art. 43 Constitución Española) obliga a ordenar desde la fabricación de los medicamentos hasta su distribución, la puesta a disposición de los mismos mediante la dispensación par parte del farmacéutico en determinados locales (oficinas de farmacia) y mediando por lo general -y en ocasiones, de manera obligatoria para el acceso a determinados medicamentos- la intervención del médico que indica a través de la receta el medicamento adecuado para la disfunción en la salud que se padece (prescripción).
Cuando se hace necesario el visado dicha medida es consecuencia de la supervisión de la prescripción por parte de la autoridad sanitaria competente con claros objetivos sanitarios y/o socioeconómicos.
jueves, 10 de noviembre de 2016
Dengue

Dengue en Paraguay
El dengue es una enfermedad infecciosa causada por un virus (Hay cuatro serotipos de virus del dengue (DEN 1, DEN 2, DEN 3 y DEN 4)). Puede adquirirse por la picadura de la hembra de un mosquito de la especie Aedes. Es común en las zonas tropicales y subtropicales. Los brotes pueden ocurrir en las épocas de lluvia.
Los síntomas aparecen transcurridos entre 3 y 14 días (promedio de 4 a 7 días) tras la picadura infecciosa.
Incluyen fiebre que puede ser alta, cefaleas, dolor retroorbitario, dolor en las articulaciones y los
músculos, vómitos y una erupción cutánea. La mayoría de las personas con dengue se recupera al cabo de 2 semanas. Hasta entonces, ingerir abundantes líquidos, reposar y tomar medicamentos contra la fiebre, distintos a la aspirina o antiinflamatorios no esteroideos, pueden ser de ayuda. Algunas veces, el dengue se convierte en fiebre hemorrágica por dengue, que causa hemorragias en la nariz, las encías o debajo de la piel. También puede evolucionar en síndrome de shock por dengue que causa hemorragia masiva. Estas formas de dengue pueden acarrear peligro para la vida del paciente.
Los síntomas aparecen transcurridos entre 3 y 14 días (promedio de 4 a 7 días) tras la picadura infecciosa.
Incluyen fiebre que puede ser alta, cefaleas, dolor retroorbitario, dolor en las articulaciones y los
músculos, vómitos y una erupción cutánea. La mayoría de las personas con dengue se recupera al cabo de 2 semanas. Hasta entonces, ingerir abundantes líquidos, reposar y tomar medicamentos contra la fiebre, distintos a la aspirina o antiinflamatorios no esteroideos, pueden ser de ayuda. Algunas veces, el dengue se convierte en fiebre hemorrágica por dengue, que causa hemorragias en la nariz, las encías o debajo de la piel. También puede evolucionar en síndrome de shock por dengue que causa hemorragia masiva. Estas formas de dengue pueden acarrear peligro para la vida del paciente.
No hay ningún tratamiento específico contra el dengue. El debgue grave es una complicación potencialmente mortal, pero su diagnóstico clínico precoz y una atención clínica cuidadosa por personal médico y de enfermería experimentado suele salvar la vida de los pacientes.

Mapa de la incidencia del dengue
Más del 70% de la carga de morbilidad por esta enfermedad se concentra en Asia Sudoriental y en el
Pacífico Occidental. En los últimos años, la incidencia y la gravedad de la enfermedad han aumentado rápidamente en Latinoamérica y el Caribe. En las regiones de África y el Mediterráneo Oriental también se han registrado más brotes de dengue en los últimos 10 años. En 2010 se notificó la transmisión indígena del dengue en dos países de Europa. Al aumento mundial del dengue han contribuido la urbanización, los movimientos rápidos de personas y bienes, las condiciones climáticas favorables y la falta de personal capacitado.
Para disminuir el riesgo, cuando viaje a países con incidencia de dengue:Pacífico Occidental. En los últimos años, la incidencia y la gravedad de la enfermedad han aumentado rápidamente en Latinoamérica y el Caribe. En las regiones de África y el Mediterráneo Oriental también se han registrado más brotes de dengue en los últimos 10 años. En 2010 se notificó la transmisión indígena del dengue en dos países de Europa. Al aumento mundial del dengue han contribuido la urbanización, los movimientos rápidos de personas y bienes, las condiciones climáticas favorables y la falta de personal capacitado.
Utilice repelentes para insectos que tengan DEET
Utilice ropa que le cubra los brazos, las piernas y los pies
Cierre las puertas y ventanas que no tengan telas protectoras
Post publicado el 22 de abril de 2013 en "Aragón y Medicina"
lunes, 7 de noviembre de 2016
Test de la Marcha

Durante la prueba se registra en cada minuto la frecuencia cardíaca, saturación de oxígeno, escala de Borg para disnea y fatiga y la aparición o no de síntomas. El 6 MWT se utiliza para conocer la evolución y calidad de vida de pacientes con enfermedades cardiorrespiratorias, ya que se considera una prueba fácil de realizar, bien tolerada, y que refleja muy bien las actividades de la vida diaria.
En la EPOC, esta prueba de esfuerzo se considera un potencial biomarcador para la gravedad de la enfermedad. Su utilidad en la patología respiratoria se encuentra influenciada tanto por el compromiso de la función respiratoria como por las manifestaciones extrapulmonares, es decir, debilidad muscular, enfermedad vascular pulmonar o depresión.
Este test se considera un importante predictor de supervivencia en los pacientes con enfermedad respiratoria crónica
jueves, 3 de noviembre de 2016
Bloqueo Auriculoventricular completo o de tercer grado

El Bloqueo Auriculoventricular completo se caracteriza por la interrupción completa de la conducción AV. Ningún estímulo generado por la aurícula pasa al ventrículo, por lo que las aurículas y los ventrículos se contraen cada uno a su ritmo
El ritmo ventricular dependerá del sitio del Sistema de Conducción donde se origine el ritmo de escape, en el Nodo AV, Haz de His o una Rama del Haz de His (mientras más alto el sitio del bloqueo, mayor Frecuencia Cardiaca y QRS más estrechos).
Si usted tiene un bloqueo cardíaco de segundo o tercer grado, los síntomas pueden incluir:
- Dolor de pecho
- Mareo
- Sensación de desmayo
- Cansancio
- Palpitaciones cardíacas. Las palpitaciones suceden cuando su corazón se siente acelerado o trepidante.
Sus características electrocardiográficas son:
Antes de indicar un Marcapasos definitivo se debe descartar causas de Bloqueo AV reversibles, como el tratamiento con fármacos bradicardizantes (betabloqueantes o antiarrítmicos) o la cardiopatía isquémica
Esta es una emergencia y requiere atención médica cuanto antes
lunes, 31 de octubre de 2016
Gastroenteritis eosinofílica

Los órganos más frecuentemente afectados son el estómago y el intestino delgado.
En la forma predominantemente mucosa la infiltración de la misma por esosinófilos produce dolor abdominal, vómitos y diarreas. En pocos casos se presenta malabsorción intestinal y pérdida de peso. En algunos casos el cuadro puede simular una apendicitis aguda.
En la forma predominantemente muscular el cuadro puede ocasionar inflamación y fibrosis, lo que puede causar un cuadro de obstrucción intestinal. En la forma subserosa los pacientes pueden presentar ascitis aislada o en combinación con los síntomas antes mencionados. En estos casos suele asociarse con más frecuencia con eosinofilia en sangre periférica y suele asociarse con una historia de alergia o intolerancia a ciertos alimentos.
En las enfermedades gastrointestinales eosinofílicas se produce inflamación y fibrosis en ausencia de desencadenante conocido, lo que se conoce como gastroenteritis eosinofílica primaria. La presencia de eosinófilos en las capas del epitelio gastrointestinal no siempre está asociada un aumento de eosinófilos en sangre periférica.
El diagnóstico de esta enfermedad es ante todo de sospecha clínica. La confirmación definitiva nos la dará el anatomopatólogo. Muchos de estos pacientes tienen una historia de asma, atopía o intolerancia a ciertos alimentos.
el diagnóstico diferencial debe realizarse con otros cuadros que produzcan síntomas intestinales y que presenten eosinofília en sangre periférica.
El tratamiento de esta enfermedad es empírico debido a los escasos estudios descritos en la literatura, los cuales son retrospectivos. En algunos casos se recomienda realizar pruebas de la IgE total y test de alergias a los principales alimentos alergénicos y evitarlos en casos de que el resultado sea positivo, junto con la necesidad de administrar dietas con aminoácidos esenciales. En el caso de que los síntomas continúen o que estos sean de gravedad se recomienda la administración de corticoides
miércoles, 26 de octubre de 2016
Tratamiento endoscópico con radiofrecuencia o HALO en el esofago de Barret

El esófago de Barrett es una lesión que afecta al revestimiento del esófago.
La enfermedad por reflujo gastroesofágico (ERGE) es un trastorno en el que el ácido del estómago y los enzimas provocan lesiones en el revestimiento del esófago, produciendo síntomas como acidez gástrica, regurgitación y dolor en el pecho. En algunos pacientes las células normales del esófago resultan dañadas y con el tiempo este daño puede dar como resultado inflamación y cambios genéticos que hacen que las células se alteren. El tejido modifica su apariencia y microscópicamente cambia («metaplasia intestinal» o esófago de Barrett). Se estima que el 13% de los pacientes con ERGE también tienen esófago de Barrett.
El diagnóstico se establece tras la realización de una gastroscopia donde se toman biopsias en la zona de inflamación esofágica. El hallazgo de células intestinales en el esófago (metaplasia intestinal) confirma el diagnóstico de esófago de Barrett. Hay distintos grados de Barrett: metaplasia intestinal, displasia de bajo grado, displasia de alto grado y adenocarcinoma esofágico.
Las recomendaciones conjuntas de las sociedades médicas indican realizar endoscopias digestivas altas con biopsia a los pacientes con esófago de Barrett, regularmente y de por vida. La frecuencia será mucho mayor en los pacientes con displasia, debido al riesgo aumentado de desarrollar un cáncer.
Además de las aproximaciones por observación endoscópica para el esófago de Barrett, existen opciones de tratamiento que incluyen la terapia endoscópica y quirúrgica para eliminar completamente el tejido de Barrett.
Entre las opciones de tratamiento endoscópico se encuentra la ablación con radiofrecuencia o HALO. Esta es una técnica en la que se calienta el tejido hasta que deja de ser viable o está vivo, mediante la utilización de una energía calorífica que se aplica de forma precisa y controlada. La tecnología de ablación HALO es capaz de conseguir la eliminación completa del tejido enfermo sin causar daños a las estructuras normales cercanas.
Actualmente hay controversia en cuanto a la utilización de la radiofreciencia en virtud de la existencia o no de displasia (vease aqui)
El tratamiento se realiza conjuntamente con endoscopia digestiva alta sin necesidad de hospitalización y no implica incisiones. Hay dos tipos de catéteres de ablación: HALO 360º y HALO 90º.
El paciente sólo pasa unas horas en el hospital. Después del procedimiento puede experimentar molestias en el pecho y dificultad para tragar durante varios días, tratándose con medicación.
Centros de la Seguridad Social que cuentan con radiofrecuencia:
- Hospital Universitario Central de Asturias.
- Hospital Marqués de Valdecilla, Santander
- Hospital de Basurto, Bilbao
- Hospital Clínic de Barcelona.
- Hospital ‘La Mancha Centro’ de Alcázar de San Juan, Ciudad Real
- Hospital Vall d’Hebrón, Barcelona
- Hospital Parc Taulí, Sabadell
- Hospital La Fe Valencia
- Hospital Virgen de Macarena Sevilla
- Hospital Clínico San Carlos, Madrid
- Hospital La Princesa, Madrid
- Hospital Gregorio Marañón, Madrid
- Hospital Gómez Ulla, Madrid
- Hospital Universitario de Bellvitge Hospitalet de Llobregat Barcelona.
- Hospital Río Hortega, Valladolid.
- Hospital Clínico Lozano Blesa, Zaragoza
- Hospital General de Ciudad Real
- Complejo Hospitalario Llenera-Zafra, Llenera (Badajoz)
- Hospital Lucus Augusti, Lugo Galicia

- Hospital Universitario Insular, Gran Canaria
- Hospital Universitario A Coruña
- Instituto Valenciano de Oncología IVO
- Hospital del Mar, Barcelona
- Hospital Universitario de Burgos
- Hospital General Valencia
- Hospital General Universitario de Alicante
- Hospital Universitario Puerto Real
- Hospital Universitari Son Espases, Palma de Mallorca
- Hospital Son Llátzer, Palma de Mallorca
- Complejo Hospitalario Toledo, Toledo
- Hospital Donostia San Sebastián, San Sebastián – Guipúzcoa
- Hospital Costa del Sol, Marbella – Málaga
- Hospital Universitari de Girona Doctor Josep Trueta, Girona
viernes, 21 de octubre de 2016
Eosinofilia pulmonar simple o Síndrome de Loeffler
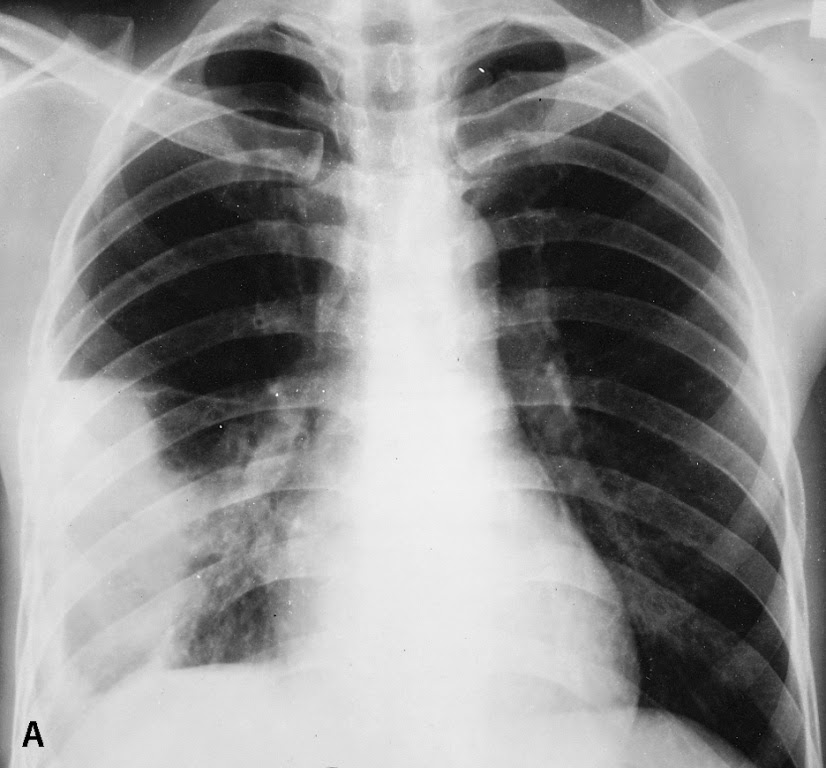
Los síntomas pueden variar desde absolutamente ninguno hasta presentarse síntomas severos (tos, fiebre, malestar general, taquicardia ...) y pueden desaparecer sin tratamiento.
Existen 3 formas posibles de diagnosticar una Eosinofilia Pulmonar: el síndrome IPE (infiltrados pulmonares y eosinofilia sanguínea), el hallazgo de eosinofilia en el LBA o la eosinofilia en el tejido pulmonar obtenido por biopsia pulmonar o transbronquial.4 La historia clínica dirigida y la exploración física son imprescindibles y deberán incluir la valoración de una ingesta previa de fármacos o productos inhalados, la posibilidad de viajes actuales o previos en zonas con endemias parasitarias.
La enfermedad con frecuencia desaparece sin tratamiento, pero si éste se necesita, la respuesta generalmente es buena. Sin embargo, se pueden presentar recaídas (la enfermedad reaparece).
martes, 18 de octubre de 2016
Ascaridiasis

Es un parásito que habita en regiones templadas y cálidas del mundo. Es mucho más frecuente en los países en vías de desarrollo donde las condiciones sanitarias son deficientes y en las áreas donde las heces humanas se utilizan como abono.
El número de infestados en el mundo se calcula que pueden llegar de 1000 a 1500 millones.
Es un nematodo cilíndrico, de color blanquecino amarillento o rosado. Está recubierto externamente por una cutícula, la epicutícula, que es una delgada pero densa película, compuesta en gran parte por lípidos.
El macho, en su estado adulto posee una longitud de 15 a 30 cm, con un diámetro de 2 a 4 mm. El extremo posterior del macho está incurvado ventralmente, y presenta un par de espículas para dilatar la vulva de la hembra y facilitar la copulación. Posee un aparato reproductor sumamente desarrollado, que ocupa casi 2/3 de la cavidad corporal del parásito. La hembra adulta mide de 25 a 35 cm de longitud y tiene un diámetro de 3 a 6 mm. Su extremo posterior es cónico. Posee un aparato reproductor muy desarrollado que, al igual que en el macho, ocupa casi la totalidad de su cuerpo. Consta de 2 ovarios filiformes, que circundan al intestino, 2 oviductos, y 2 úteros que se unen y continúan con la vagina. La vagina desemboca en la vulva, en el 1/3 anterior de la cara ventral del cuerpo del parásito. El aparato digestivo está formado por la boca con tres labios finamente dentados; estos dentículos son visibles con scanning y son diferentes en Áscaris suum y Áscaris lumbricoides. El esófago se continúa con el intestino, y el recto desemboca en la cloaca sexual en el macho, y en el ano en la hembra.
El tamaño de los huevos, varía de 55 a 75 um, la mayoría son ovalados de color ambarino. Pueden encontrarse de diversas formas, aquellos que presentan una corteza en toda la superficie y son elongados, son conocidos como infértiles y los que únicamente presentan una parte interna que muestra claramente la producción de una larva y su corteza externa y la membrana interna son bien claras, son los fértiles.
El hombre se infesta al consumir huevos larvados o embrionados de Áscaris lumbricoides.
Cuando se ingieren los huevos y éstos llegan al intestino, eclosionan y se convierten en larvas. Las larvas empiezan entonces a desplazarse por el cuerpo.
Una vez atraviesan la pared intestinal, las larvas pasan del hígado a los pulmones a través del torrente sanguíneo. Durante esta etapa, pueden aparecer síntomas pulmonares como la tos (incluso se pueden expulsar lombrices al toser). En los pulmones, las larvas ascienden por los bronquios hasta la garganta, donde son tragadas. Luego regresan al intestino delgado, donde crecen, maduran, se reproducen y ponen huevos. Las lombrices alcanzan la madurez aproximadamente 2 meses después de que la persona ingiera los huevos.
Las lombrices adultas viven y permanecen en el intestino delgado. Una lombriz hembra puede producir hasta 240.000 huevos al día, que se eliminan a través de las heces y luego entran en un período de incubación que dura varias semanas. Los niños son particularmente susceptibles a contraer ascaridiasis porque tienden a meterse cosas en la boca, incluyendo tierra, y sus hábitos higiénicos suelen ser más deficientes que los de los adultos.
Los reservorios son el hombre y el cerdo.
Las fuentes de infestación del ascariasis son principalmente el suelo, los alimentos, agua, manos y objetos contaminados con materia fecal que contiene huevos de Áscaris lumbricoides.
El período de transmisibilidad ocurre durante el estadio de huevo embrionado con larva infestante y luego de 2 o 3 semanas de la defecación con huevos.
El contagio no se da de persona a persona.
Aunque la infección helmíntica puede no dar ningún síntoma, cuanto mayor es la cantidad de lombrices implicadas, más graves son los síntomas que padece la persona afectada. Los niños tienen más probabilidades que los adultos de desarrollar síntomas gastrointestinales porque sus intestinos son más pequeños y presentan un riesgo más elevado de desarrollar obstrucción intestinal.
Los síntomas de una infestación leve son:
Lombrices en las heces
Tos con expulsión de lombrices
Pérdida de apetito
Fiebre
Respiración sibilante.
en los casos más graves pueden aparecer síntomas más preocupantes:
Vómitos
Dificultad para respirar
Distensión abdominal (hinchazón abdominal)
Fuertes dolores abdominales
Obstrucción intestinal, vólvulos e incluso apendicitis
Obstrucción de las vías biliares (que incluyen el hígado y la vesícula biliar)
Se sospecha por los síntomas antes referidos y se confirma por la expulsión del parásito o un examen de heces que demuestre la presencia de huevos.
El tratamiento consiste en la administración de medicamentos antiparasitarios por vía oral para eliminar las lombrices intestinales. A veces las heces se vuelven a analizar aproximadamente 3 semanas después de aplicar el tratamiento para determinar si contienen huevos y/o lombrices. En contadas ocasiones es necesario extirpar quirúrgicamente las lombrices (particularmente en los casos de obstrucción intestinal, obstrucción de las vías hepáticas o infección abdominal). Si el niño tiene ascaridiasis, le deberán hacer pruebas para determinar si tiene otros parásitos intestinales como, por ejemplo, oxiuros.
Por lo general, los síntomas suelen desaparecer durante la primera semana de tratamiento.
Para evitar posibles reinfestaciones, es recomendable adoptar las siguientes precauciones:
• Asegúrese de que el niño se lava bien las manos, especialmente después de utilizar el sanitario y antes de las comidas.
• Lleve regularmente a los animales domésticos que convivan con la familia al veterinario para que éste compruebe si tienen lombrices.
• Mantenga las uñas del niño cortas y limpias.
• Esterilice cualquier prenda de ropa contaminada, como los pijamas y la ropa de cama.
• Evalúe el origen de la infestación. Podría ser necesario adoptar medidas higiénicas adicionales dentro o alrededor de su casa.
La medida de protección más importante contra la ascaridiasis es desechar de forma segura e higiénica los excrementos humanos, que pueden transmitir huevos. En los lugares donde se utilizan las heces humanas como abono, se deben cocinar muy bien todos sus alimentos y limpiarlos con una solución de yodo apropiada (particularmente las frutas y hortalizas).
A los niños adoptados procedentes de países en vías de desarrollo se les suelen practicar pruebas de cribado para la detección de lombrices como medida de precaución. A los niños que viven en áreas subdesarrolladas se les puede recetar medicación antiparasitaria como tratamiento preventivo. Las siguientes prácticas son recomendables para todos los niños:
• Intente en la medida de lo posible evitar que su hijo se meta cosas en la boca.
• Enseñe a su hijo a lavarse las manos a conciencia y frecuentemente, especialmente después de usar el váter y antes de las comidas.
miércoles, 5 de octubre de 2016
El Chagas en España

Aproximadamente dos tercios de los afectados son mujeres entre los 30 y 40 años, y hasta el 90 % podrían proceder de Bolivia.
Los indices de detección y tratamiento de la Enfermedad de Chagas, enfermedad tropical considerada como desatendida, son todavia muy bajos en España a pesar de que ya se dispone de instrumentos diagnósticos y terapéuticos
por el que se obliga a todos los centros de transfusión a realizar estudio serológico a todo posible donante nacido, hijo de madre nacida o que haya sido transfundido en países donde la enfermedad es endémica.
Desde 2005 se controla la vía de trasmisión por vía sanguinea a través del cribado en los bancos de sangre y de trasplantes de órganos. Además, el 14 de Marzo de 2008 se publicó el Plan Nacional de Sangre de Cordón, donde se recogen las mismas recomendaciones que con la transfusión sanguínea para todo potencial donante. Sin embargo, la detección de una posible transmisión vertical es un tema pendiente de regulación.
España todavia no cuenta con ningún programa nacional de control de esta enfermedad, solo algunas comunidades autónomas (Cataluña, Comunidad Valenciana) tienen un protocolo específico, por lo que sería necesario establecer un protocolo común en España.
lunes, 3 de octubre de 2016
El desfibrilador como tratamiento precoz en las arritmias malignas ventriculares

El objetivo inicial se basa en la prevención, es decir, impedir o disminuir las posibilidades de una muerte súbita llevando a cabo los reconocimientos periodicos que las sociedades médicas establecen.
Pero ocurre que no en todos los estudios se detecta una afección cardiaca por lo que resulta fundamental conocer el tratamiento adecuado.
La estimulación cardiaca mediante un desfibrilador representa una forma de tratamiento eficaz para las arritmias ventriculares por mecanismo de reentrada en el ritmo eléctrico cardiaco. La utilización de los DEA o DESA en los primeros minutos hace que la supervivencia aumente considerablemente.
El desfibrilador externo semiautomatico (DESA) puede resultar fudamental en las instalaciones deportivas o incluso en puntos estrategicos donde se acumule mucha personal (espacios cardioprotegidos). Se hace necesario asegurar la formación a todo el personal susceptible de utilizar los desfibriladores.
Uso del DESA
miércoles, 21 de septiembre de 2016
El Mal de Alzheimer, una enfermedad familiar

La demencia tipo Alzheimer es una enfermedad neurodegenerativa que cursa con deterioro cognitivo y trastornos en la conducta, incapacitando de manera considerable a la persona que lo padece además de afectar al entorno más cercano del individuo: la familia.
Se calcula que uno de cada diez personas mayores de 65 años padecen la enfermedad.
En la mayoría de los casos, algunos miembros de la familia adoptan el papel de ‘cuidador’ y se ven
atrapados por la enfermedad.
El cuidador, en una mayoría de ocasiones, ha de estar pendiente del enfermo las 24 horas del día y durante 365 días al año, lo que supone una dependencia absoluta por la carga y sufrimiento emocional al no saber cómo reaccionar ante ciertas situaciones, provocando un anticipo de duelo hacia la pérdida. Esta situación, debido a la falta de recursos y que incluye la hospitalización, visitas médicas, residencias o fármacos y que en un porcentaje elevado son asumidos por la familia, lleva al cuidador a abandonar su vida laboral. A nivel social se produce un aislamiento y se le dedica menos tiempo al resto de los miembros de la familia y/o amistades.
Es fácil que el cuidador caiga en la idea errónea de pensar que puede cuidar al enfermo porque lo conoce, dando por hecho que el cuidado también va a ser mejor que el que se facilita en los centros geriátricos o especializados Hay que hacer ver al cuidador que, si es posible, llegado el caso, debe poner al enfermo en manos de profesionales que sepan tratar a estos enfermos. Hay que convencer a los cuidadores de que deben proteger su salud mental.
El mal de Alzheimer es uno de los retos sociales y sanitarios más importantes que se encuentran en la
sociedades industrializadas cara a las próximas décadas.
A continuación se hace mención a 15 puntos clave para cuidar a un enfermo de Alzheimer
1. Seguir una rutina diaria. Asegurarse de que haya muchos objetos familiares alrededor.
2. Mantenerse al tanto de dónde está el paciente y de su seguridad. Un método que algunas familias usan para prevenir que el paciente se extravíe es ponerle campanas a todas las puertas que den al exterior.
3. Asegurar que el paciente coma bien y beba una abundancia de líquidos.
4. Ayudar al paciente a que se mantenga los más independiente posible por el mayor tiempo posible.
5. Proporcionar oportunidades para que el paciente haga ejercicios regularmente y no se olvide de la
recreación.
6. Seguir relacionándose con los amigos y la familia.
7. Emplear ayudas escritas para la memoria como calendarios y grandes relojes, listas de las tareas diarias, recordatorios acerca de las rutinas o medidas de seguridad e identificando las etiquetas en los objetos que pueden olvidarse. Actualmente existen aplicaciones informáticas como las que se pueden encontrar en http://stimuluspro.com/
8. Asegurarse que el paciente se haga chequeos regulares.
9. Planificar necesidades futuras como la atención en Centros de Día y el ingreso en una Residencia.
10. Dar mucho apoyo emocional al paciente y a todos los prestadores de asistencia.
11. Asegurarse que el paciente tome los medicamentos regularmente, si se prescriben.
12. Asociarse a un grupo de apoyo para los miembros de la familia.
13. Pedir ayuda con las finanzas, arreglos legales, asesoramiento diario, temas emocionales, atención de reposo o arreglos del hogar para convalecientes cuando se necesite.
14. Revisar el hogar en cuestiones de medidas de seguridad, como barras en la pared cerca de la taza del baño y la bañera, luces nocturnas en los pasillos y en las escaleras, alfombras no resbaladizas, etc.
15. Asegurarse que todos los profesionales sanitarios tengan una lista completa de todas las recetas y todos los medicamentos sin receta del paciente.
Comunicarse con una persona con Alzheimer
Suscribirse a:
Entradas (Atom)